Lab & Testing Services
- Phone:
- 416-813-7200
- Hours:
- Customer service representatives are available Monday to Friday, 7 a.m. to 5 p.m. EST.
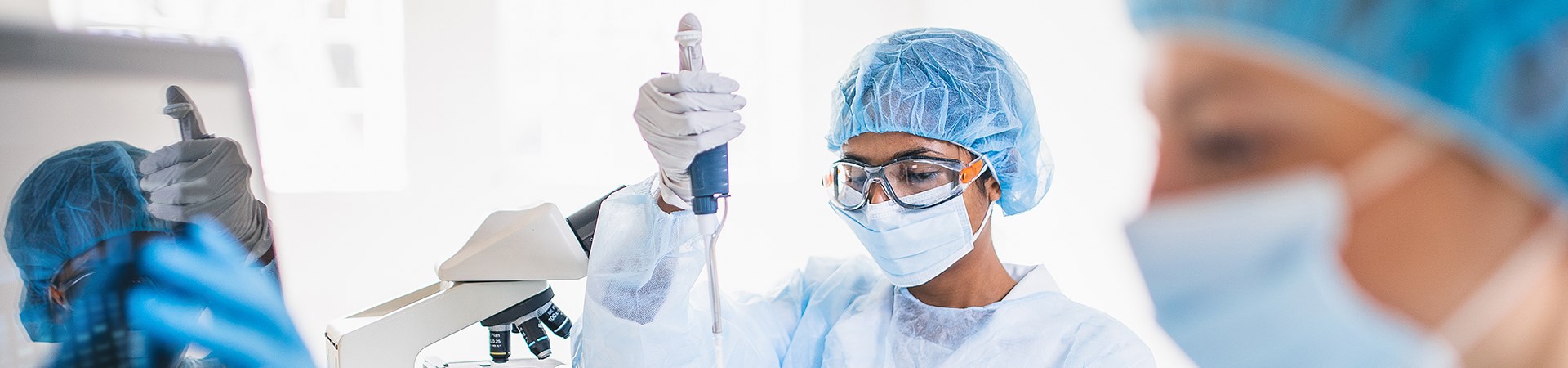
Welcome to the Department of Paediatric Laboratory Medicine
The Department of Paediatric Laboratory Medicine (DPLM) at SickKids is a leading, world-class facility working towards reducing the impact of childhood illness by quickly providing diagnoses that lead to early selection of best treatment options. DPLM is a valuable and integral part of the SickKids team, generating original research and providing timely, specialized diagnostic services for health-care providers in Canada and around the world.
The department includes five divisions: Clinical Biochemistry, Genome Diagnostics, Haematopathology, Microbiology, Pathology.
Requisitions
Download requisition forms.
Test Catalogue
Browse our test catalogue.

Dr. Gino Somers MBBS, BMedSci, PhD, FRCPA
Chief, Paediatric Laboratory Medicine
Professor, Department of Laboratory Medicine and Pathobiology, University of Toronto
Dr. Somers is a paediatric pathologist who is internationally recognized for his work in paediatric sarcomas, test development and medical leadership. His research has led to a better understanding of the drivers of paediatric cancer and improved cancer diagnostics. He is currently working to improve faculty and trainee wellness at Hospital and University levels.

Laboratory Divisions
Clinical Biochemistry
The Division of Clinical Biochemistry is known as a reference paediatric clinical biochemistry laboratory offering comprehensive and specialized laboratory services and technical/scientific expertise worldwide. The laboratory has considerable expertise in dealing with small sample volumes and relevant analytical methods suitable for the analysis of paediatric specimens.

Head, Division of Clinical Biochemistry
Routine biochemistry tests are available upon request for all biological fluids including whole blood, serum, plasma, urine, CSF and other fluids. Please contact the lab manager for further information.
The comprehensive Therapeutic Drug Monitoring (TDM) laboratory is able to analyze a broad range of drugs utilizing advanced techniques to help clinicians and pharmacists provide effective and safe drug therapies to individual patients.
The Toxicology laboratory is able to analyze a wide range of specimens for close to 1,500 drugs and chemicals. With our direct connection to the Ontario Poison Centre and the American Association of Poison Centers we are able to quickly develop new tests that are current with changing drug trends.
The Metabolic Diseases laboratory is dedicated to service, education and research in the diagnosis and treatment of inborn errors of metabolism. The majority of these disorders present in childhood and patients are often initially diagnosed with some other condition.
Laboratory personnel have a high level of technical and interpretive skills and are equipped to handle a broad spectrum of disorders with emphasis on:
- Amino acidurias
- Organic acidurias
- Glycogen storage and fatty acid oxidation disorders
- Lysosomal storage diseases
We are certified as a Tay-Sachs screening centre (NTSAD) and participate in ERNDIM and CDC international quality assurance programmes. We offer prenatal diagnosis of certain conditions.
The Mitochondrial Laboratory offers a wide range of mitochondrial functional enzyme tests that aid in diagnosis and monitoring of children with mitochondrial genetic diseases. These functional assays are unique as they assess the functional impact of specific mitochondrial genetic mutations and their potential consequences in disease presentation and clinical outcome.
The CALIPER project is aimed at developing pediatric reference intervals that can be used to identify abnormal test results in children with medical concerns. This will enable physicians to more accurately and efficiently diagnose and treat children.
Genome Diagnostics
The Division of Genome Diagnostics specializes in genetic testing for rare hereditary disorders affecting the paediatric population. The Division includes the Cytogenetic Laboratory, Molecular Genetics Laboratory, Microarray and a clinical fibroblast service.
Head, Division of Genome Diagnostics
The Genome Diagnostics Laboratory offers clinical genetic testing for rare genetic conditions, with a focus on those that affect the paediatric population. We are staffed by professionally trained and certified molecular geneticists, genetic counsellors, molecular technologists, as well as genome analysts, bioinformaticians and technicians.
The laboratory offers several test services including:
- disease-specific assays
- targeted testing for familial variants
- targeted panels for recurrent disease-causing variants
- multi-gene panels
- genome-wide sequencing*
*The Genome Diagnostics Laboratory is a partner in Genome-wide Sequencing Ontario (GSO); a pilot implementation project for rare disease diagnostics. GSO provides clinical whole exome and whole genome analysis in Ontario.
Our staff work closely with clinicians to facilitate appropriate genetic testing for patients and assist in the interpretation of results and their relevance to clinical care. For inquiries, please contact us at molecular.lab@sickkids.ca
The Cytogenetics Laboratory specializes in the genomic analysis of paediatric specimens, referred for a wide range of:
- constitutional conditions
- cancer diagnosis and management
We are a Children’s Oncology Group (COG)-Approved laboratory. The laboratory is unique in the province of Ontario in having a substantial caseload of specialized testing for the chromosome instability syndromes.
The Cytogenetics Laboratory accepts specimens for genomic microarray testing submitted with a referral of developmental delay and/or multiple congenital anomalies. For more information, please see the Microarray Info Sheet (pdf). Specimens submitted to SickKids for genomic microarray testing must be accompanied by the Genomic SNP Microarray Test Requisition.
Genomic microarray analysis may be indicated if a diagnosis is not achieved after G-band analysis. Genomic microarray analysis will not detect polyploidy, genomic imbalances of regions that are not represented on the microarray, and low-level mosaicism.
If an abnormality is identified by microarray, peripheral blood specimens for follow up FISH analyses of the patient and parents will be requested in the microarray report to assist with interpretation of the results. For follow-up FISH analyses of abnormal microarray results, specimens should be submitted along with the Constitutional Analysis Requisition.
Requirements and recommendations
- Patients must have a valid Ontario Health Card number.
- Pre-test counseling of patients and their families is recommended.
- Genomic microarray analysis is not recommended for patients with infertility or multiple miscarriages; G-band analysis should be requested for these indications.
- G-band/FISH analysis should continue to be requested as the front-line test for suspected aneuploidy syndromes including Down syndrome, Trisomies 13 and 18, and sex chromosome aneuploidy, or for patients with suspected chromosomal mosaicism.
The Clinical Fibroblast Service (CFS) is a biorepository service that establishes, banks and distributes fibroblast cell lines, primarily for diagnostic testing.
All requests must be accompanied by a signed requisition completed appropriately for the required service(s).
Note: Services for research fibroblast and lymphoblastoid cell lines are available at The Centre of Applied Genomics.
Acceptable Specimen Types
Fibroblast cell lines can only be established from live cells. Acceptable specimens for this service are limited to fresh tissue biopsy (not frozen or fixed) originating from skin, cartilage, umbilical cord or other body-part containing connective tissue from which fibroblast lines could be generated.
Details About Our Service (PDF) Instructions for Submission (PDF)
Haematopathology
The Division of Haematopathology includes the following laboratories. Basic and collaborative research in the Division is focused on hematologic disorders of children.

Head, Division of Haematopathology
The General Haematology Laboratory, provides a 24 hour, seven days a week essential service for the performance of CBCs, blood film and bone marrow examination.
The Special Haematology Laboratory provides tests for the diagnosis of thalassemia, haemoglobinopathies and red cell enzyme defects.
The Coagulation Laboratory provides service for the diagnosis and management of patients with haemophilia, vonWillebrands, and other hereditary and acquired hemorrhagic and thrombotic disorders.
The Flow Cytometry Laboratory provides testing for the diagnosis and monitoring of children with hematological malignancies and immunological disorders.
The Molecular Haematopathology Laboratory detects the presence of the chromosomal translocations that have implications on the clinical course of childhood leukemia.
The Blood Transfusion Laboratory is responsible for the manipulation and distribution of all blood products. Tests performed include routine compatibility testing and special immunohematology tests.
The stem cell laboratory is responsible for processing and issue of haematopoietic stem cell products. The tissue laboratory processes and stores heart valves and pericardium allografts required for cardiac surgery programs.
Microbiology
The Division of Microbiology has played an important role over the past 60 years in many significant developments in the diagnosis and pathogenesis of childhood infectious diseases, still the dominant cause of morbidity and mortality in infancy and childhood.
Head, Division of Microbiology
Lab services
At the present time, the laboratory provides diagnostic services for a wide range of infections in children, from neonates, to healthy children with common infections, to the severely immunocompromised host (transplant, HIV patient) with both common and unusual bacterial, fungal, viral and parasitic infections.
Complementing the main diagnostic sections of Bacteriology, Virology, Serology and Mycology, a comprehensive Molecular Microbiology section provides cutting edge rapid diagnostics for children with infectious diseases. The laboratory is also very active in teaching and research, with strong connections to the University of Toronto.
Pathology
Paediatric Pathology is a distinct discipline that links the fundamental molecular and cellular abnormalities underlying disease processes in children to the clinical signs and symptoms of those diseases. Using techniques such as histology, cytology, morphometry, enzyme histochemistry, immunochemistry, in situ hybridization, electron microscopy and molecular biology, the pathologist pursues the diagnosis and pathogenesis of these important diseases in children.
Head, Division of Pathology
Our division performs hospital-consented autopsies, and provides a consultation service for the examination of post-mortem tissues including referred-in cases from the Coroner’s Office or outside hospitals, glass slides, organ blocks, or referral of fetuses or neonatal infants for a complete post-mortem examination. We provide an efficient and comprehensive consultation service to hospitals throughout Ontario, and will review selected cases from outside of this geographic area. Information about the procedure for handling specific cases is available.
Our electron microscopy facility has the capability to perform highly specialized testing and their applications to paediatric diseases, including examination of metabolic and storage diseases, examination of cilia, renal biopsy evaluation, muscle biopsy evaluation, scanning electron microscopy evaluation of hair specimens for variations caused by genetic abnormalities, evaluation of congenital enteropathies (microvillous inclusion disease, e.g.), urinary stone analysis and specialized research techniques such as cryoimmobilization and immunogold labeling.
We provide histology services which include consultations and histotechnology techniques on surgical, cytological, autopsy, veterinary and industrial research materials. This involves processing, embedding, microtomy and/or staining of paraffin-embedded and frozen tissues with a variety of specialized histochemical stains, many of which are specialized pediatric stains (acetylcholinesterase for the diagnosis of Hirschsprung disease, e.g.).
Immunohistochemistry and most recently in situ hybridization has contributed greatly to modern diagnostic pathology due to its specificity and sensitivity. For the last two decades, we have developed a comprehensive range of antibody markers and probes for immunohistochemical diagnostic service, many of which are pediatric-specific, and we develop new antibody tests regularly.
The molecular pathology laboratory is currently expanding its test menu and incorporating several new technologies to aid in the diagnosis, prognosis and therapeutic decision making in pediatric cancers. In addition to PCR and RTPCR-based assays we now offer SNP/microarray testing of tumour tissue for medulloblastoma, low-grade glioma and neuroblastoma, which allows for the detection of gene copy number changes and variants; NanoString® assays for medulloblastoma (subtyping into the four main molecular categories) and sarcoma (detection of diagnostically specific fusion transcripts). In collaboration with molecular hematopathology, we are using a NanoString® assay for the detection of specific fusion genes in pediatric leukemia.
We perform morphometric analysis on glass slides, photomicrographs (negatives or prints), transmission electron micrographs, and digitized images. This includes consultations, training and analysis on all types of materials, including surgical, autopsy and research specimens. This involves capturing images from raw materials, counting and measuring cells and tissue structures, plotting histograms and/or graphs, and providing final statistical analysis aided by our computerized systems
The Neuropathology Laboratory specializes in handling surgical, autopsy and research specimens for neurosurgery, neurology, ophthalmology, orthopedics and plastic surgery.
Our expertise includes the following procedures: muscle biopsies and muscle histograms, nerve biopsies, cryotomy/cryosections, Golgi silver preparation, enzyme histochemical staining techniques, as well as processing, embedding, microtomy and staining of paraffin embedded tissue and frozen tissue. We also provide consultation and hands-on-training for most techniques including immunohistochemistry.

To discuss research projects or other related studies, please contact the Division Head of the respected area.
Through its affiliation with the SickKids Research Institute and The Centre for Applied Genomics, the Genome Diagnostics division is committed to supporting research and translating new genetic discoveries and technologies into clinical care for patients and their families. Particular research interests by the Division Head, Dr. Somerville include familial enteropathy; health impact of hereditary cardiac disease management and implementation of high-resolution, low-cost diagnostics.
Basic and collaborative research in the Division of Haematopathology is focused on hematologic disorders of children. Other research interests include cell death and RNA helicases.
The Division of Pathology has a strong independent and collaborative research program investigating many different aspects of childhood disease including paediatric sarcomas utilizing diagnostic molecular pathology, and many of our researchers have received international recognition for their work.
Research within the Division of Microbiology largely focus on (but not limited to):
- Respiratory viral infections
- Invasive fungal infections
- Cystic fibrosis microbiology
- Clostridium difficile infections in children
- Standards of practice for diagnostic pediatric microbiology
- Molecular microbiology as applied to pediatric infectious disease

Members of DPLM contribute many hours of teaching to University of Toronto undergraduate, graduate and professional students, as well as The Michener Institute for Applied Health Sciences technology students.
Graduate student supervision is a key responsibility of many members in the department. The practice of laboratory medicine is taught within accredited programs in all the divisions. In addition, educational programs in laboratory medicine, as it applies to clinical disciplines, exist for areas such as general paediatrics, clinical genetics, haematology/oncology and infectious diseases.
In collaboration with The Michener Institute for Applied Health Sciences, St. Lawrence College and Ontario Tech University, DPLM has an active educational program for technicians/technologists in training, providing a unique opportunity to train future technologists in laboratory medicine as applied to paediatrics. DPLM members also reach out to the community, sharing their particular expertise in the diagnosis and management of childhood illnesses.
Each division within DPLM is very active in both graduate and postgraduate teaching through its affiliation with the University of Toronto and house a number of basic and clinically-oriented research projects (supported by numerous grants from national and provincial granting foundations).
The training activities of the Division in Paediatric Clinical Chemistry span every level of post-secondary education and the program is among the largest in Canada.
Uniquely tailored programs have been devised for:
- Trainees in Medical Laboratory Technology from the Michener Institute
- Fellows in the Postdoctoral Training Program in Clinical Chemistry and medical residents in Endocrinology from the University of Toronto
- Clinical Pharmacy Residents
- Clinical Pharmacology Fellows
- Genetic Metabolic Fellows
- Canadian College of Medical Genetics Fellows
- Several other medical specialties
Recent development of a Fellowship Program funded by the Sanford Jackson Endowment has allowed us to train postdoctoral fellows in Paediatric Clinical Biochemistry. These include the recent international visiting scientists and clinical biochemists from various countries.
The Genome Diagnostics division is the accredited training centre for the Fellowship of the Canadian College of Medical Geneticists for Molecular Genetics and Cytogenetics.
The Microbiology division supports training programs for laboratory technologists, laboratory technicians, medical microbiology and pediatric infectious diseases residents. Research interests include viral respiratory infections, the microbiology of cystic fibrosis, the study of invasive fungal infections and related rapid diagnostics, Clostridium difficile infections in children, standards of practice for diagnostic paediatric microbiology, and the applications of molecular microbiology to paediatric infectious diseases.
The Pathology division offers a fellowship in paediatric anatomic pathology for a one-year appointment, renewable for a second year. The first year provides comprehensive experience in paediatric autopsy and surgical pathology. Fellows participate in resident teaching and clinical conferences, and are encouraged to undertake a research project. For those desiring a second year, there is the opportunity for more extended research experience and training in molecular techniques as applied to the diagnosis of paediatric disease.
Requirement: MD degree or certification or Board eligibility in pathology.
Stipend: Dependant on level of training.
Pathology’s educational activities cover a broad range of students, including laboratory technologists, laboratory technicians, pathologists’ assistants, pathology and neuropathology residents, and clinical fellows, as well as undergraduate teaching at the University and national webinars.
In the Division of Pathology, a wide spectrum of paediatric disorders is included annually in the 6,000 surgical specimens and approximately 250 neonatal and paediatric autopsies, including forensic cases. There are active biopsy programs in liver, kidney, gut, heart and neuromuscular disorders.
The Division is staffed by eight pathologists and four scientists and has advanced facilities for electron microscopy, immunohistochemistry and molecular pathology. In addition to a full complement of paediatric specialties SickKids boasts an active and integrated Research Institute. There are well-established department research programs for a number of paediatric disorders, including SIDS, the molecular basis of childhood neoplasia, and renal, lung, neural, cardiac and gastrointestinal diseases.
Teaching awards
The PLM Senior Scientist Teaching Award, first presented in 1997, highlights the enormous value the Paediatric Laboratory Medicine places on excellence in teaching.
In 2007, a new award, DPLM Technologist Teaching Award, was created to recognize the considerable teaching activities of DPLM technologists. The awards are an opportunity to recognize colleagues who have extensively contributed to the field of education as teachers, course coordinators and mentors. The nominees will have established themselves as inspirational role models.
Award recipients are selected from nominations made by their PLM peers, with awards being presented at the DPLM Annual Laurence E. Becker Symposium.
To nominate a peer, DPLM staff are invited to send a short synopsis of why they believe their nominee should be recognized with one of the awards.
Licensing and accreditation
DPLM has successfully participated in and has received many accreditation certifications provincially, nationally and internationally. Expand the sections below to view current certificates and licenses.
MOH Lab Licence 2022–2024 (pdf)
Expiry: October 29, 2024
Accreditation Canada ISO 15189 Plus 2023–2027 (pdf)
Expiry: March 19, 2027
Health Canada - CTO Tissue (pdf)
Expiry: December 31, 2023
Foundation for the Accreditation of Cellular Therapy (FACT) (pdf)
Expiry: January 24, 2024
Health Canada - CTO - Lymphohematopoietic Cells (pdf)
Expiry: December 31, 2023
College of American Pathologists (CAP) (PDF)
Expiry: June 18, 2024
Centre for Medicare & Medicaid Services: Clinical Laboratory Improvement Amendments (PDF)
Expiry: August 17, 2024
- American College of Medical Genetics (ACMG)
- Canadian Immunohistochemistry Quality Control for Pathology (CIQC)
- CDC Newborn Screening Program
- Charles River Proficiency Test Program
- College of American Pathologists (CAP)
- Continuous Improvement and Qualifying for Accreditation Project (CIQAP)
- Euroimmune - Serology
- European Research Network for evaluation and improvement of screening, Diagnosis and treatment of Inherited Disorders of Metabolism (ERNDIM)
- Exchange with Mount Sinai
- FHCRC - Otsuka Pharmaceuticals and Seattle Cancer Care Center (Peer group assessment program)
- Immprove Quality Assurance Scheme - Immunology
- Institute for Quality Management in Healthcare (IQMH)
- Institute for Research Information and Quality Assurance
- International External Foundation Quality Assessment Programme (ECAT)
- International Proficiency Testing Schemes (IPTS)
- International Tay-Sachs Testing (NTSAD)
- Internal Split Sample Procedure (I.S.S)
- KKGT - Stichting Kwaliteitsbewaking Klinische Geneesmiddelanalyse en Toxicologie (Association for Quality Assessment in Therapeutic Drug Monitoring and Clinical Toxicology) - Netherlands
- Labquality EQA Program
- LGC Standards
- National Lab for HIV Immunology
- National Lab for HIV Reference and Service (NLHRS)
- National Microbiology Laboratory – Molecular Microbiology
- North American Specialized Coagulation Laboratory Association (NASCOLA)
- Pharmacokinetic
- Post analytical DNA Sequencing Challenge SEC
- Qnostic – Molecular Microbiology
- Quality Control for Molecular Diagnostics (QCMD)
- Siemens Urinalysis Proficiency Study
- Stichting Kwaliteitsbewaking Medische Laboratoriumdiagnostiek (Dutch: Foundation for Quality Medical Laboratory Diagnostics) - (SKML)
- United Kingdom National External Quality Assessment Service (UKNEQAS)
- Vitamin D External Quality Assessment Scheme (DEQAS)
Statement of responsibility
Outlined below are the standard terms and conditions which apply to the referral of all specimens for laboratory testing to SickKids unless otherwise stipulated in a separate agreement.
Responsibility of SickKids
As a referral laboratory, under Institute for Quality Management in Healthcare (IQMH) standards, SickKids is responsible for:
- Reporting test results including critical/significant results to the referring health care institution. (Genetics results will be reported directly to the ordering physician).
- Providing documented or verbal interpretation of results.
- Complying with all applicable legal requirements to protect the privacy of all patient information.
- Notifying the referring health care institution immediately if we suspect non-proficient performance which may affect test results.
Respiratory Virus Watch Newsletter
The Respiratory Virus Watch Newsletter summarizes results of testing patient specimens for influenza and other viral respiratory pathogens at the Microbiology Laboratory. View the current issue as well previous issues here.
Annual Laurence E. Becker Symposium - Advances in Paediatric Laboratory Medicine
In affiliation with the Department of Pathobiology and Laboratory Medicine at the University of Toronto, DPLM offers an annual educational program on recent advances in paediatric laboratory medicine. Since 2003, the symposium has been named after Dr. Laurence E. Becker, the founding Chief of Paediatric Laboratory Medicine.
The symposium features national/international speakers and is attended by laboratorians and other health-care providers from across Ontario.
The format of the symposium includes presentation by keynote speakers in the morning, followed by poster sessions in the afternoon. Awards are presented for the best posters in a number of categories, including graduate students, fellows, clinical residents and technologists.
This education event is approved as an Accredited Group Learning Activity under Section 1 of the Framework of CPD options for the Maintenance of Certification, RCPSC.
Our founding chief: Dr. Laurence E. Becker
March 1943 - July 2002
Laurence E. Becker was the Founding Chief of the Department of Paediatric Laboratory Medicine (DPLM) at The Hospital for Sick Children (SickKids) and Professor, Laboratory Medicine and Pathobiology and Paediatrics at the University of Toronto. He spent his entire career at SickKids, which began in 1972 as a resident in Neuropathology.
Laurence (Larry) Becker graduated from the University of Alberta Medical School in 1967. In 1974, following a postgraduate specialization in neuropathology at the University of Toronto and at the Johns Hopkins Medical School in Baltimore, he joined the staff at SickKids as a neuropathologist. He was appointed Professor, Department of Laboratory Medicine & Pathobiology and Paediatrics at the University of Toronto in 1980, and Pathologist-In-Chief at SickKids in 1994. In 1996, after 22 years at SickKids, he became the first Chief of DPLM.
At SickKids, Dr. Becker’s passion was evident in his leadership of DPLM, and in the diagnostic work he carried out behind the scenes. As Chief of DPLM, Dr. Becker never tired of explaining and arguing for the importance of paediatric laboratory medicine and the specialized knowledge that is required to support an effective diagnostic service for children.
Dr. Becker was considered a world-leading authority on childhood neuropathology. He will be particularly remembered for advancing knowledge of paediatric cerebral tumours and sudden infant death syndrome. Dr. Becker held research grants from a wide variety of Canadian and international sources. He was an invited speaker at scientific meetings around the world and received many awards and honours. He authored more than 325 peer-reviewed publications in scientific journals, 44 book chapters, and more than 300 brief communications.
Dr. Becker founded the DPLM Symposium in 1997 to showcase advances in paediatric laboratory medicine and highlight achievements of the amalgamated department. Since 2003, the Symposium has been named ‘Laurence E. Becker Symposium’ in honour of Dr. Becker’s outstanding contribution to SickKids and the medical profession. This annual symposium is supported in part by the Laurence Becker Fund through SickKids Foundation.
Contact information and hours
Phone
Toll Free: 1-855-381-3212
Local: 416-813-7200
Customer service representatives are available Monday to Friday, 7 a.m. to 5 p.m. EST.
Shipping address
The Department of Paediatric Laboratory Medicine
The Hospital for Sick Children
170 Elizabeth Street
Room 3642 (Rapid Response Lab)
Toronto, Ontario
M5G 1H3
Laboratory operating hours
- Biochemistry: 7 days/week, 24 hours/day
- Haematology & Coagulation: 7 days/week, 24 hours/day
- Therapeutic Drug Monitoring and Toxicology: 7 days/week, 24 hours/day
- Clinical Test Development: Monday to Friday, 8 a.m. to 4 p.m.
- Metabolic Disease: Monday to Friday, 8 a.m. to 4 p.m.
- Mitochondrial: Monday to Friday, 9 a.m. to 5 p.m.
- Molecular Haematopathology: Monday to Friday, 8:30 a.m. to 5:30 p.m.
- Flow Cytometry: Monday to Friday, 8 a.m. to 5 p.m
- Microbiology (including Molecular Microbiology, Serology, Virology): Monday to Friday, 8 a.m. to 7 p.m.; Saturday, Sunday and holidays, 9 a.m. to 5 p.m
- Genome Diagnostics: Monday to Friday, 8 a.m. to 5 p.m.
- Cytogenetics: Monday to Friday, 8 a.m. to 5 p.m.
Specimen drop off hours and locations
For drop-offs after 7 p.m. please only use the 170 Elizabeth Street entrance, the University entrance is locked. All samples can be dropped off 24 hours a day in the Rapid Response Lab, Room 3642.
Rapid Response Lab
Includes Biochemistry, Therapeutic Drug Monitoring and Toxicology Laboratory, Clinical Test Development, Metabolic Disease Laboratory, Haematology, Coagulation, Molecular Haematopathology, Mitochondrial
Hours: 7 days/week, 24 hours/day
Location: Atrium, Room 3642
Directions: Take Atrium elevators to the 3rd floor, walk west (away from the glass wall at the end of the building) and follow the signs to the Rapid Response Lab.
- Toll Free: 1-855-381-3212
- Local: 416-813-7200
Customer service representatives are available Monday to Friday, 7 a.m. to 5 p.m. EST.
Microbiology
Includes Molecular Microbiology, Serology, Bacteriology
Hours: Monday to Friday, 8 a.m. to 7 p.m.
Saturday, Sunday and holidays 9 a.m. to 5 p.m.
Location: Atrium, Room 3676
Phone: 416-813-6000
Clinical Fibroblast Service
Hours: Monday to Friday, 8 a.m. to 5 p.m.
Location: Black Wing, Room 3415
Phone: 416-813-7654 ext. 302394
Email: clinicalfibroblastservice.requests@sickkids.ca
Genome Diagnostics (Molecular Genetics & Cytogenetics)
Hours: Monday to Friday, 8 a.m. to 4:30 p.m.
Location: Black Wing, Room 3416
Phone: 416-813-7200 ext. 2
Email Molecular Lab: molecular.lab@sickkids.ca
Email Cytogenetics: cytogenetics.requests@sickkids.ca
Customer service representatives are available Monday to Friday, 8 a.m. to 5 p.m. EST.
Pathology
Hours: Monday to Friday, 8 a.m. to 5 p.m.
Location: Burton Wing, Room 3141
Phone: 416-813-5967